Archives
- June 2022
- May 2020
- June 2016
- March 2016
- January 2016
- December 2015
- November 2015
- September 2015
- August 2015
- July 2015
- June 2015
- May 2015
- April 2015
- March 2015
- February 2015
- January 2015
- December 2014
- November 2014
- October 2014
- September 2014
- August 2014
- July 2014
- June 2014
- May 2014
- April 2014
- March 2014
- February 2014
- January 2014
- December 2013
- November 2013
- October 2013
- September 2013
- August 2013
- July 2013
- June 2013
- May 2013
- April 2013
- March 2013
- February 2013
- January 2013
- December 2012
- November 2012
- October 2012
- September 2012
- August 2012
- July 2012
- June 2012
- May 2012
- April 2012
- March 2012
- February 2012
- January 2012
- December 2011
- November 2011
- October 2011
- September 2011
- August 2011
- July 2011
- June 2011
- June 2010
- May 2010
Categories
- Aging & Dementia
- Alcohol related dementia
- Alzheimer's and Dementia
- Coronavirus (COVID-19) and Alzheimer's
- Creutzfeldt-Jacob Dementia
- Dementia Care
- Dementia news
- Dementia Today
- Frontotemporal Dementia
- Huntington’s Disease
- Lewy body dementia
- Parkinson's Disease Dementia
- Vascular dementia
- Wernicke-Korsakoff Syndrome
Meta
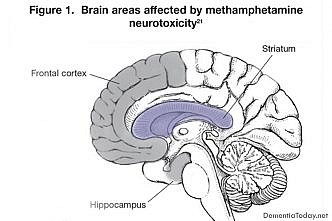
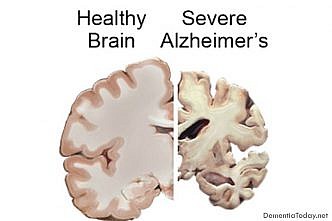
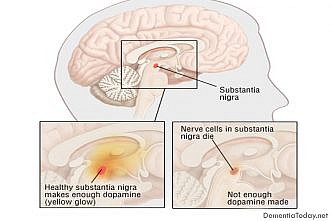
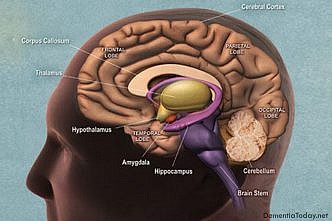
All The Latest News

COVID Brain Changes Show Parallels With Alzheimer's Disease
A study from researchers at Columbia University Vagelos College of Physicians and Surgeons reports that the brains […]

Covid-19 pandemic disrupts clinical trials in Alzheimer’s disease
The pharmaceutical industry has been as affected as any other industry by the Covid-19 pandemic; most countries’ […]

Coronavirus: Information for people affected by dementia
During the coronavirus pandemic we have advice and practical tips for people living with dementia and those […]
Caring for Someone with Alzheimer’s During the COVID-19 Outbreak: 5 Tips
Demonstrating how to wash your hands for a loved one with Alzheimer’s disease can help ensure […]

COVID-19: Advice for caring for people with Alzheimer’s disease, mild cognitive impairment
With the COVID-19 pandemic at the forefront of daily life, how much information should you share with […]

Coronavirus (COVID-19): Tips for Dementia Caregivers
Most likely, dementia does not increase risk for COVID-19, the respiratory illness caused by the new coronavirus, […]

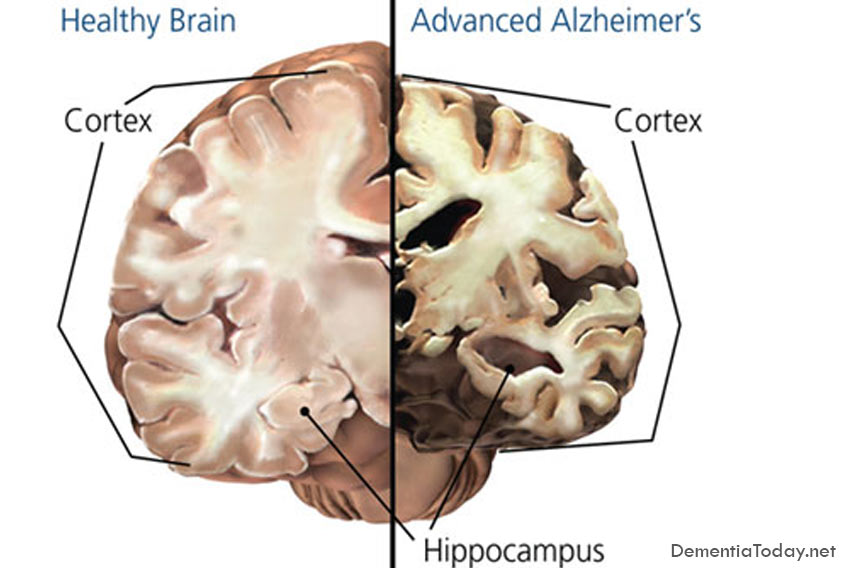
Alzheimer's Disease Dementia
Alzheimer's disease (AD) is the seventh leading cause of all deaths in the United States and […]
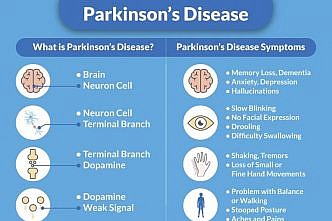
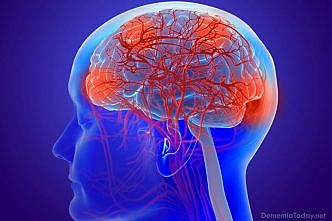
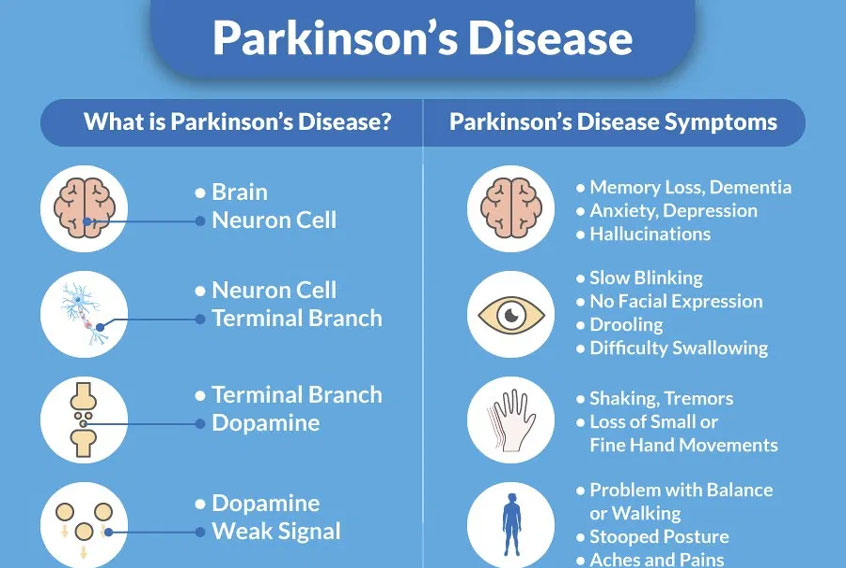
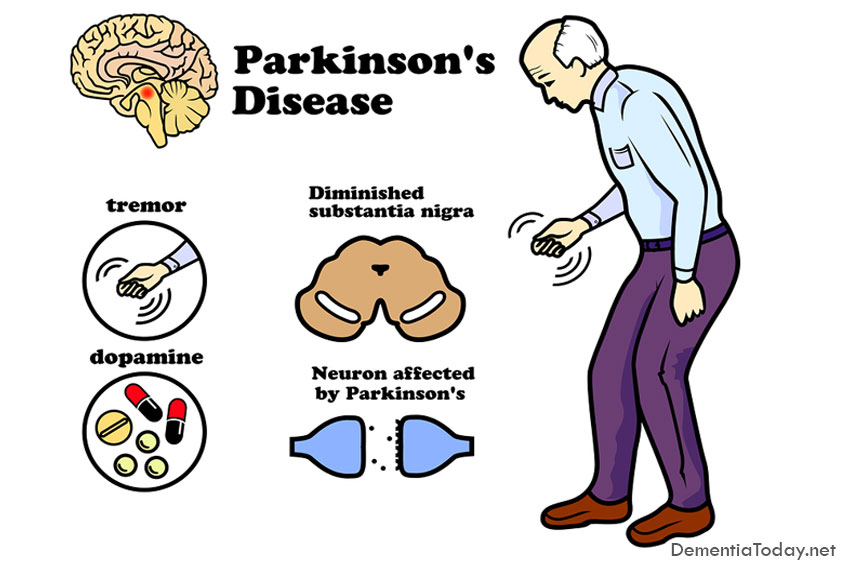
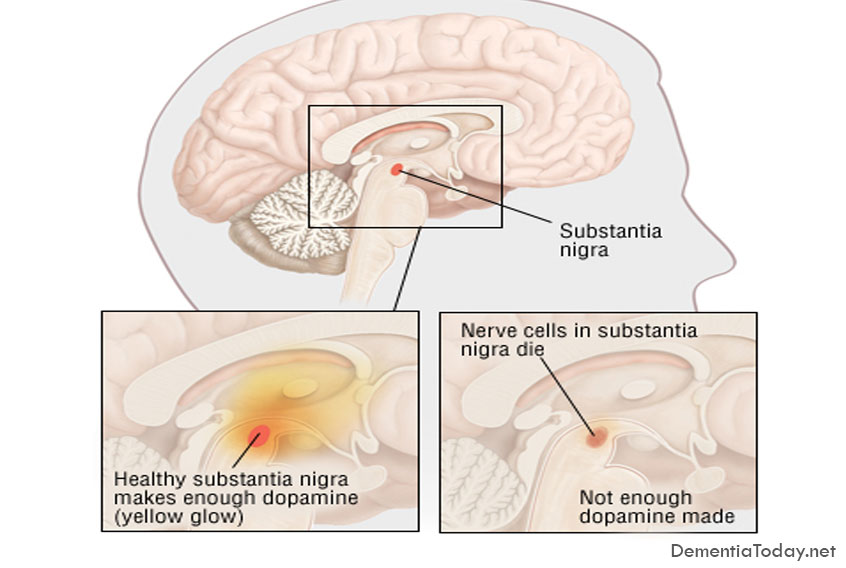
Parkinson's Disease Dementia
Parkinson's dementia is a condition that some people can experience as their Parkinson's progresses. It […]
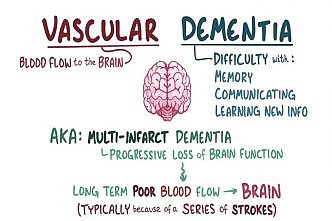
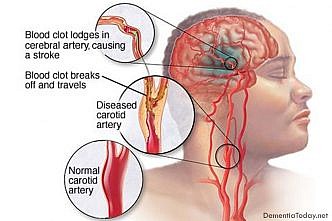
Vascular dementia
Vascular dementia is one of the most common forms of dementia, ranking only second to […]
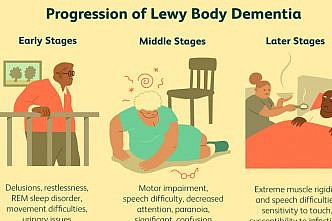
Lewy body dementia
Definition Lewy body dementia, the second most common type of progressive dementia after Alzheimer's disease, causes […]
New link found between diabetes and Alzheimer’s disease
Drugs used to treat diabetes could also be used to treat Alzheimer's disease, and vice versa, according […]
Alzheimer’s researchers find clues to toxic forms of amyloid beta
Much of the research on Alzheimer's disease has focused on the amyloid beta protein, which clumps together […]
Editor's Choices

- Advertisement -
Latest In Categories
-

We’ve been in business for 18 years and are Derbyshire trusted Psychiatry authority for 5 years! Derby Psychiatry Association, Inc., the company that owns and operates Dementia Today (for 1+ years), has researched the Psychiatry industry independently since 1992.
Recent Posts



Copyright © 1995 - 2020, Dementia Today.net, Derby Psychiatry Association, Inc.. All rights reserved
Contact Us | Sitemap |About Us | Terms & Conditions | Privacy Policy | Advertise with us